

If you are building machine learning software that informs, drives, or replaces clinical decisions, you are building a Software as a Medical Device (SaMD) under FDA jurisdiction. This is not a theoretical regulatory concern -- it determines whether your product can legally be marketed in the United States, what documentation you must produce, and what happens when your model needs to be updated.
Yet most engineering teams encounter FDA requirements late in the development cycle, typically when a regulatory consultant reviews their near-complete product and produces a list of documentation gaps that require months of remediation. The clinical AI models that ship fastest are those where engineering teams understand FDA requirements from the start and build compliance into their MLOps pipeline architecture.
This guide is written for engineers, not regulatory professionals. It explains what SaMD is, what the FDA actually requires for AI/ML devices, how Predetermined Change Control Plans work, and what documentation you need to produce. It includes a compliance checklist, a PCCP template structure, and real examples of cleared AI/ML devices.
What Is SaMD and Does Your Product Qualify?
The International Medical Device Regulators Forum (IMDRF) defines SaMD as software intended to be used for one or more medical purposes that perform those purposes without being part of a hardware medical device. The FDA adopted this definition and applies it to software that:
- Diagnoses a disease or condition (e.g., AI that reads chest X-rays for pneumonia)
- Treats or mitigates a disease (e.g., closed-loop insulin dosing algorithms)
- Screens or monitors patients (e.g., continuous arrhythmia detection)
- Aids in clinical decision-making (e.g., sepsis risk prediction displayed to clinicians)
Software that does not qualify as SaMD includes: EHR systems (administrative), clinical communication tools (messaging), practice management software, and general wellness apps without disease-specific claims.
The critical distinction is the intended use statement. A model that predicts sepsis risk and displays it to clinicians is SaMD. The exact same model used internally for hospital capacity planning (without clinical use) may not be SaMD. Your intended use statement, not your technology, determines regulatory classification.
The FDA's AI/ML Action Plan

The FDA published its Artificial Intelligence/Machine Learning Action Plan in January 2021 and has refined it through multiple guidance documents since. The framework recognizes that AI/ML software is fundamentally different from traditional medical devices because it learns and changes. Key components include:
Total Product Lifecycle (TPLC) Approach
Traditional medical devices are validated once at clearance. AI/ML devices change over time as they are retrained on new data. The FDA's TPLC approach evaluates not just the current model but the manufacturer's ability to manage model changes safely over the product's entire lifecycle.
Good Machine Learning Practice (GMLP)
In October 2021, the FDA, Health Canada, and the UK's MHRA jointly published 10 GMLP principles. These are the foundational requirements for any AI/ML medical device. Here is what each principle means for engineering teams:
| Principle | What It Requires | Engineering Implication |
|---|---|---|
| 1. Multi-disciplinary expertise | Development team includes clinical, data science, and regulatory expertise | Document team composition and each member's qualifications in the design history file |
| 2. Good software engineering practices | Version control, testing, CI/CD, design controls | IEC 62304 software lifecycle process; traceability from requirements to tests |
| 3. Representative clinical study participants | Training and validation data reflects the intended patient population | Document demographics of training data; analyze and report any underrepresentation |
| 4. Independent training and test datasets | Strict separation of data used for training, validation, and testing | Log dataset splits with hashes; ensure no data leakage between partitions |
| 5. Best available reference datasets | Ground truth labels must be clinically validated | Document labeling methodology, inter-annotator agreement, and label quality metrics |
| 6. Model design tailored to data | Model complexity should match data availability | Justify model architecture choice relative to dataset size; document hyperparameter selection rationale |
| 7. Focus on performance of human-AI team | Evaluate how humans interact with AI outputs | Conduct usability testing with clinicians; measure human+AI performance, not just model accuracy |
| 8. Testing demonstrates clinically valid performance | Performance metrics must be clinically meaningful | Report sensitivity, specificity, PPV, NPV -- not just AUROC; use clinical thresholds agreed with clinician stakeholders |
| 9. Security and reliability | Device must be resilient to cyberattack and operational failure | Threat modeling, adversarial testing, graceful degradation design, cybersecurity documentation |
| 10. Monitoring deployed models | Continuous post-market monitoring of real-world performance | Implement drift detection and performance tracking in production |
Predetermined Change Control Plans (PCCPs)
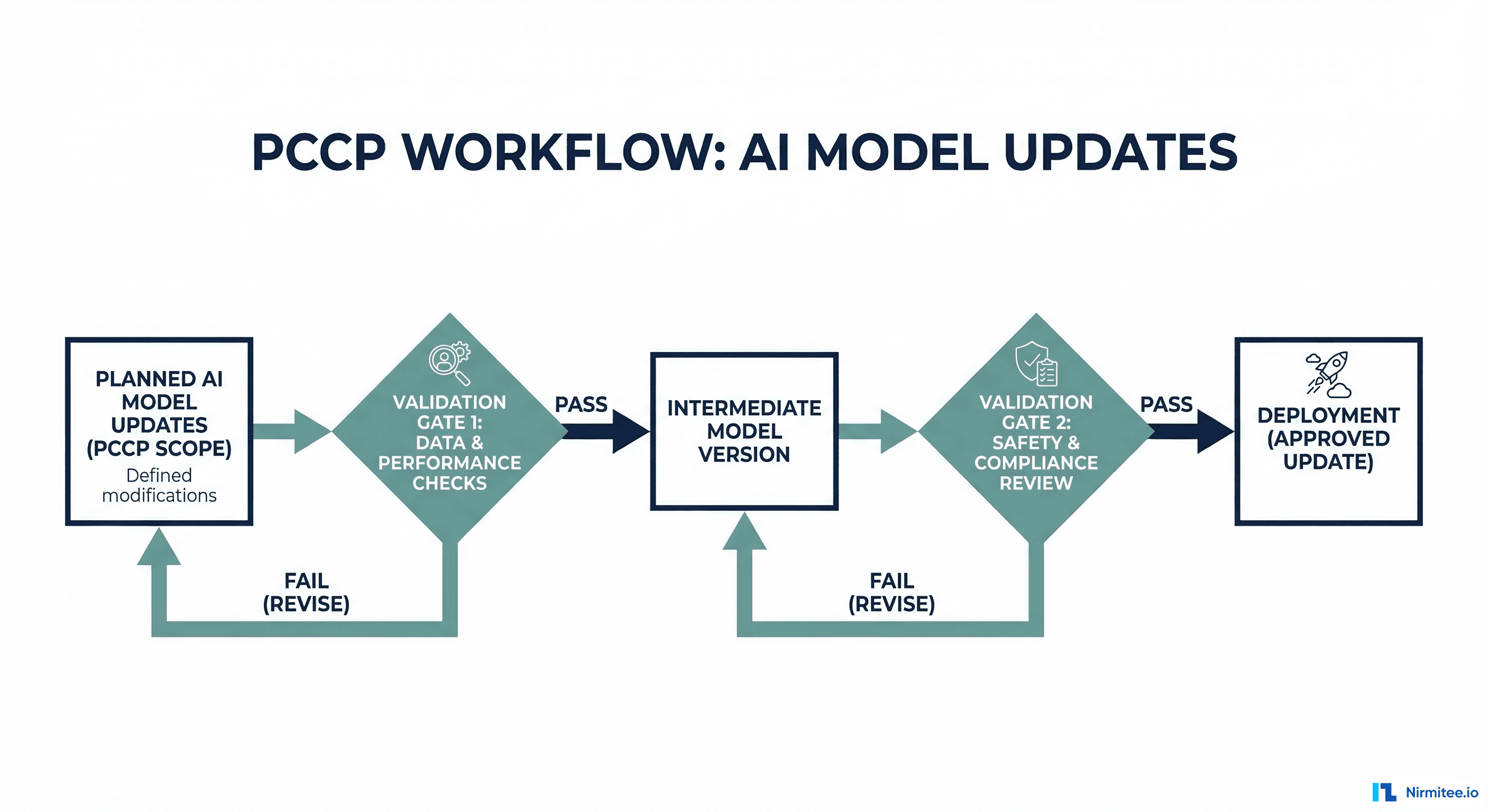
PCCPs are the most significant regulatory innovation for AI/ML devices. They allow manufacturers to describe, in advance, how their model will change after initial clearance -- and get those changes pre-approved as part of the original submission. Without a PCCP, every model update (retraining on new data, adjusting thresholds, adding features) requires a new regulatory submission.
What a PCCP Must Include
The FDA's September 2024 final guidance on PCCPs specifies four required components:
1. Description of Modifications: What types of changes are planned? Retraining on new data? Threshold adjustments? Feature additions? Architecture changes? Each type must be explicitly described. Vague statements like "the model may be updated" are insufficient.
2. Modification Protocol: How will each change be implemented? This must describe the retraining methodology, data requirements, validation protocol, and deployment procedure in enough detail that the FDA can evaluate whether the process reliably produces safe updates.
3. Impact Assessment: How will the manufacturer verify that each change maintains safety and effectiveness? This requires specifying: performance thresholds the updated model must meet, statistical tests to compare pre- and post-update performance, fairness analysis across demographic groups, and clinical validation criteria.
4. Labeling Changes: How will device labeling be updated to reflect modifications? If retraining changes the model's performance characteristics (e.g., improved sensitivity at the cost of specificity), the labeling must be updated to reflect this.
PCCP Template Structure
Here is a practical template structure for engineering teams preparing a PCCP submission:
predetermined_change_control_plan:
document_version: "1.0"
device_name: "SepsisAlert Pro"
submission_type: "510(k) with PCCP"
planned_modifications:
- id: "MOD-001"
type: "Periodic Retraining"
description: |
Retrain the sepsis prediction model on accumulated
production data to address data drift.
trigger: |
PSI exceeds 0.25 on any critical feature, OR
rolling 30-day AUROC drops below 0.85.
frequency: "As needed, estimated quarterly"
- id: "MOD-002"
type: "Threshold Adjustment"
description: |
Adjust the alert threshold to optimize sensitivity/
specificity tradeoff based on clinical feedback.
trigger: "Clinical review committee recommendation"
range: "Alert threshold between 0.5 and 0.9"
- id: "MOD-003"
type: "Feature Addition"
description: |
Add new lab values or vital signs as input features
when clinical evidence supports their predictive value.
constraints: |
Maximum 5 new features per update cycle.
Each feature must have published clinical evidence.
modification_protocol:
data_requirements:
minimum_samples: 10000
minimum_positive_rate: 0.05
demographic_representation:
- "Age groups: 18-44, 45-64, 65-84, 85+"
- "Sex: Male, Female"
- "Race: White, Black, Hispanic, Asian, Other"
recency: "At least 60% from most recent 6 months"
training_procedure:
architecture: "Frozen (same as cleared model)"
hyperparameters: "Same search space as original"
cross_validation: "5-fold stratified"
reproducibility: "Fixed random seed, logged config"
validation_procedure:
holdout_set: "20% of data, stratified, time-based split"
metrics:
- name: "AUROC"
threshold: ">= 0.85"
comparison: "Non-inferior to cleared model"
- name: "Sensitivity (at 90% specificity)"
threshold: ">= 0.70"
- name: "Calibration slope"
threshold: "Between 0.8 and 1.2"
fairness_analysis:
method: "Equalized odds across demographic groups"
threshold: "AUROC disparity less than 0.05 between groups"
deployment_procedure:
stages:
- "Shadow deployment (14 days minimum)"
- "Canary deployment to single unit (14 days minimum)"
- "Full rollout with monitoring"
rollback_criteria: |
Performance drops below validation thresholds
during shadow or canary period.
impact_assessment:
safety_monitoring:
- "Track false negative rate (missed sepsis cases)"
- "Monitor alert fatigue via alert-to-action ratio"
- "Compare patient outcomes pre/post update"
reporting: "Quarterly performance report to clinical committee"Risk Classification for AI/ML Devices
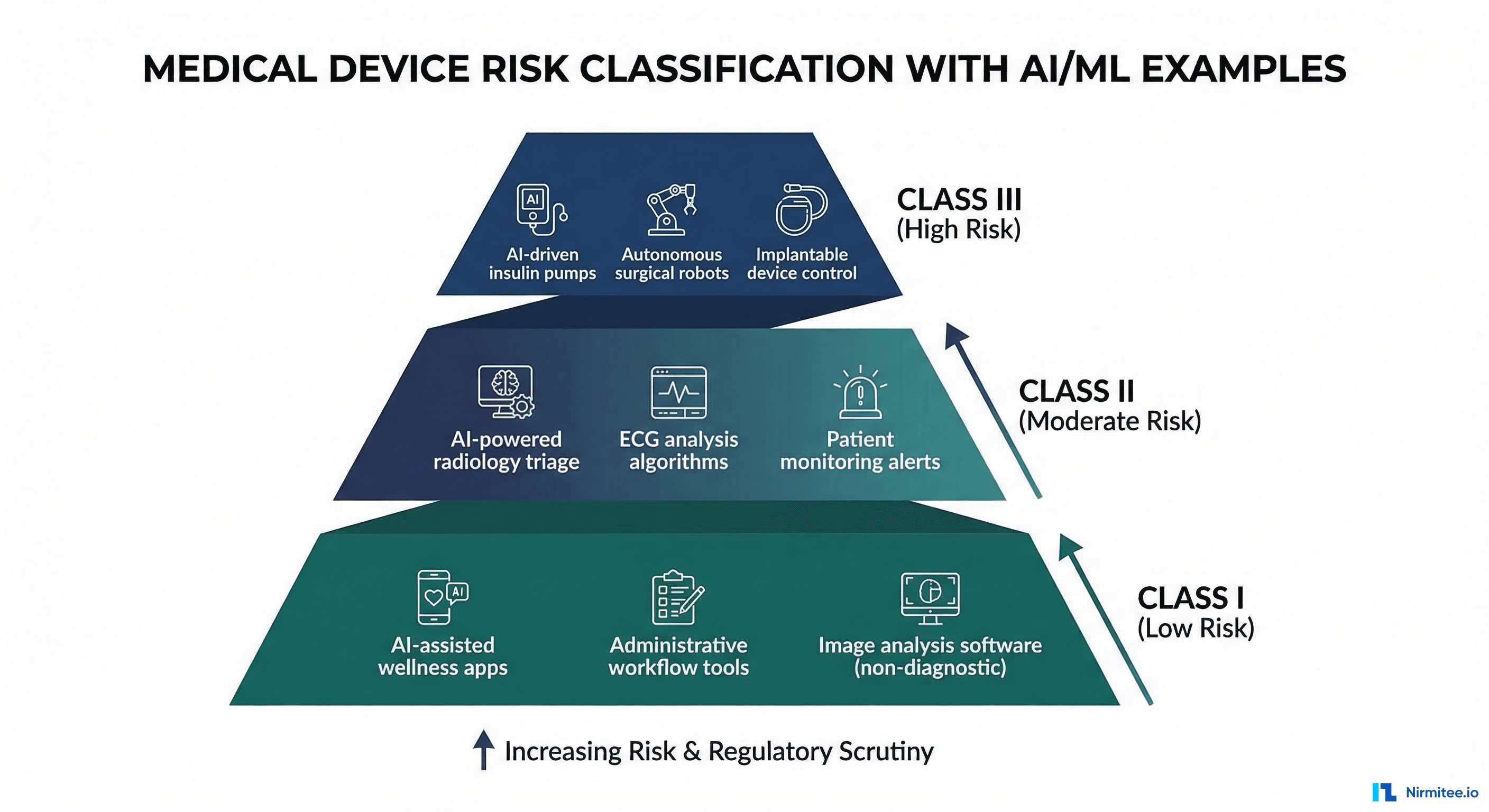
The FDA classifies medical devices into three risk classes. The classification determines the regulatory pathway and the level of evidence required for clearance.
| Class | Risk Level | Regulatory Pathway | AI/ML Examples | Documentation Level |
|---|---|---|---|---|
| Class I | Low risk | 510(k) exempt (most cases) | Clinical workflow tools, non-diagnostic AI assistants | General controls only |
| Class II | Moderate risk | 510(k) or De Novo | Sepsis prediction, diabetic retinopathy screening, ECG interpretation, radiology AI | General + special controls, performance testing |
| Class III | High risk | Premarket Approval (PMA) | Closed-loop drug delivery, autonomous surgical AI, life-sustaining algorithms | Full clinical trial data, PMA submission |
The vast majority of current AI/ML devices are Class II, cleared through the 510(k) or De Novo pathway. As of early 2026, the FDA has cleared over 1,000 AI/ML-enabled medical devices, with approximately 85% classified as Class II.
510(k) vs De Novo: Choosing the Right Pathway
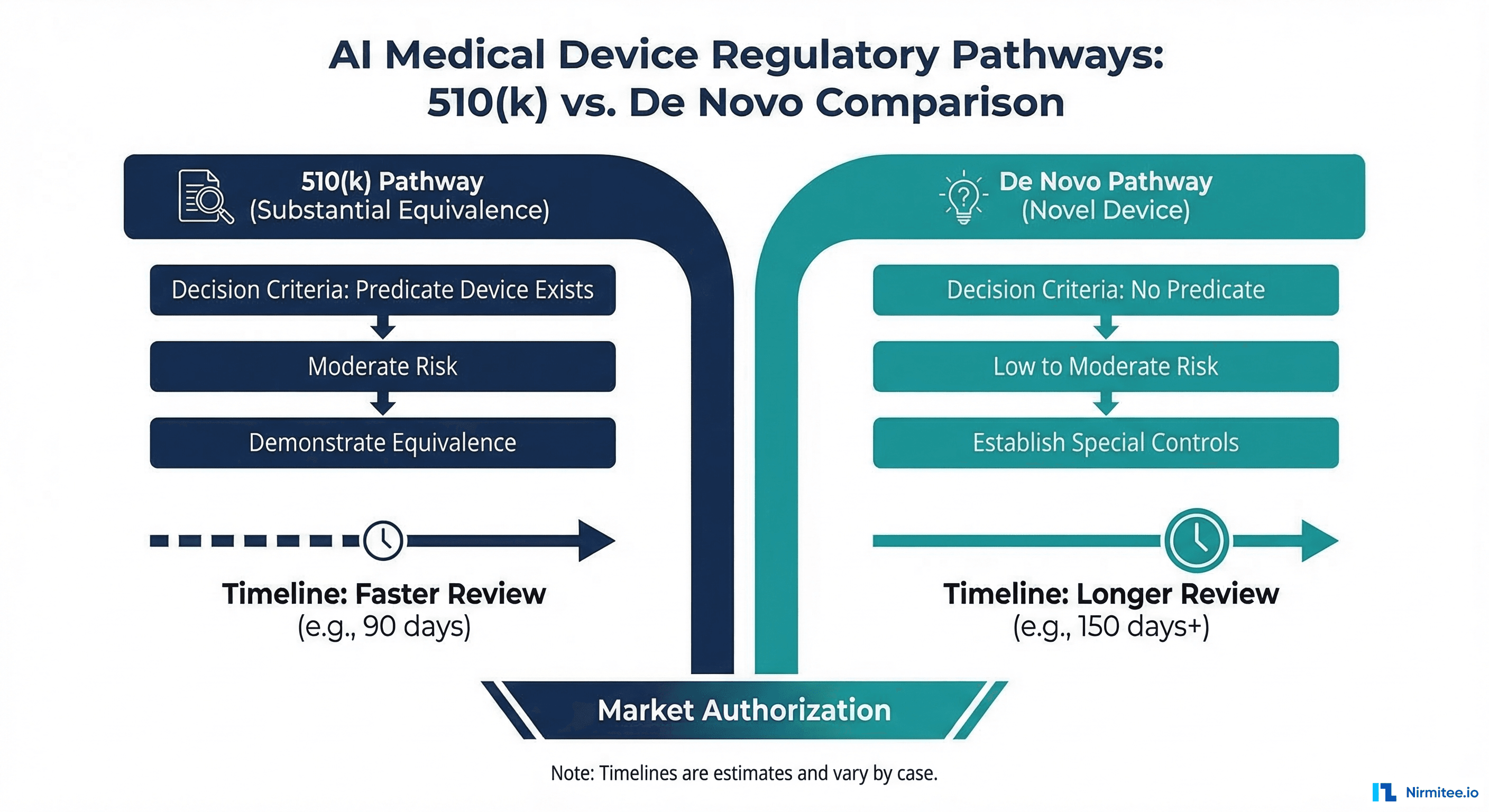
For Class II AI/ML devices, two regulatory pathways exist:
510(k) Pathway
The 510(k) pathway requires demonstrating that your device is substantially equivalent to a legally marketed predicate device. You must identify a predicate (an already-cleared device with similar intended use and technology) and show that your device performs at least as well.
Advantages: faster review (typically 90-120 days), lower evidence burden, established precedent for many AI device categories. Disadvantages: requires a suitable predicate device, which may not exist for truly novel AI applications.
De Novo Pathway
The De Novo pathway is for novel, low-to-moderate risk devices without a predicate. You must demonstrate safety and effectiveness through performance testing and propose the special controls that should apply to your device category. Once granted, your De Novo becomes a predicate for future 510(k) submissions by other manufacturers.
Advantages: no predicate required, you define the product category. Disadvantages: longer review (typically 150-300 days), higher evidence burden, more extensive documentation.
Decision framework: Search the FDA's AI/ML device database for cleared devices with similar intended use. If a suitable predicate exists, pursue 510(k). If your device is genuinely novel (new clinical application, new AI approach without precedent), pursue De Novo.
Documentation Requirements for Engineers

The FDA does not prescribe exact documentation formats, but the following artifacts are expected in any AI/ML device submission. Engineers should produce these as part of the development process, not as retrospective documentation exercises.
Data Documentation
- Data source description: Where did the training data come from? What institutions, what time period, what EHR systems?
- Data collection protocol: How was data selected? What inclusion/exclusion criteria were applied?
- Demographic analysis: Age, sex, race/ethnicity, and geographic distribution of the training population
- Labeling methodology: How were ground truth labels determined? Who labeled the data? What was the inter-annotator agreement?
- Data split documentation: Exact methodology for train/validation/test split, with dataset hashes for reproducibility
- De-identification verification: Evidence that PHI was removed from training data (Safe Harbor checklist or Expert Determination report)
Algorithm Documentation
- Architecture description: Model type, layer configuration, number of parameters, input/output specification
- Feature engineering: Complete list of input features with clinical rationale for each
- Hyperparameter selection: Search methodology, final values, and rationale
- Training procedure: Loss function, optimizer, learning rate schedule, early stopping criteria, training duration
- Software dependencies: Complete list of libraries and versions (requirements.txt or environment.yml)
Performance Documentation
- Primary performance metrics: AUROC, sensitivity, specificity, PPV, NPV at the operating threshold
- Subgroup analysis: Performance disaggregated by age group, sex, race/ethnicity, and disease severity
- Calibration analysis: Calibration plot, Brier score, calibration slope and intercept
- Comparison to predicate (510(k)): Head-to-head performance comparison on the same test set
- Failure mode analysis: Known limitations, edge cases, and populations where performance may be reduced
from dataclasses import dataclass, field
from typing import List, Dict, Optional
import json
from datetime import date
@dataclass
class ModelCard:
"""FDA-aligned model documentation for AI/ML medical devices."""
# Device identification
device_name: str
model_version: str
intended_use: str
indications_for_use: str
submission_type: str # "510(k)", "De Novo", "PMA"
# Data documentation
training_data_sources: List[str] = field(default_factory=list)
training_data_size: int = 0
training_data_date_range: str = ""
demographic_distribution: Dict[str, Dict[str, float]] = field(default_factory=dict)
labeling_methodology: str = ""
inter_annotator_agreement: float = 0.0
# Algorithm documentation
model_architecture: str = ""
num_parameters: int = 0
input_features: List[Dict[str, str]] = field(default_factory=list)
training_config: Dict = field(default_factory=dict)
# Performance documentation
test_set_size: int = 0
primary_metrics: Dict[str, float] = field(default_factory=dict)
subgroup_metrics: Dict[str, Dict[str, float]] = field(default_factory=dict)
calibration_metrics: Dict[str, float] = field(default_factory=dict)
known_limitations: List[str] = field(default_factory=list)
# PCCP reference
pccp_version: Optional[str] = None
last_update_date: Optional[str] = None
update_history: List[Dict] = field(default_factory=list)
def generate_report(self) -> str:
"""Generate FDA-formatted model documentation report."""
report = {
"device_identification": {
"name": self.device_name,
"version": self.model_version,
"intended_use": self.intended_use,
"submission": self.submission_type
},
"data_management": {
"sources": self.training_data_sources,
"total_samples": self.training_data_size,
"date_range": self.training_data_date_range,
"demographics": self.demographic_distribution,
"labeling": self.labeling_methodology,
"iaa_score": self.inter_annotator_agreement
},
"algorithm_description": {
"architecture": self.model_architecture,
"parameters": self.num_parameters,
"features": self.input_features,
"training": self.training_config
},
"performance_assessment": {
"test_set": self.test_set_size,
"metrics": self.primary_metrics,
"subgroups": self.subgroup_metrics,
"calibration": self.calibration_metrics,
"limitations": self.known_limitations
}
}
return json.dumps(report, indent=2)
# Example usage
model_card = ModelCard(
device_name="SepsisAlert Pro",
model_version="2.1.0",
intended_use="Aid ICU clinicians in early identification of sepsis",
indications_for_use="Adult patients (18+) in ICU settings",
submission_type="510(k) with PCCP",
training_data_sources=["Hospital A (2022-2025)", "Hospital B (2023-2025)"],
training_data_size=85000,
primary_metrics={
"auroc": 0.89,
"sensitivity_at_90spec": 0.78,
"ppv": 0.34,
"npv": 0.98
}
)Real Examples of Cleared AI/ML Devices
Understanding what has already been cleared helps calibrate expectations for your own submission. Here are notable cleared AI/ML devices across categories:
| Device | Manufacturer | Function | Pathway | Year |
|---|---|---|---|---|
| IDx-DR | Digital Diagnostics | Autonomous diabetic retinopathy diagnosis | De Novo | 2018 |
| Viz.ai ContaCT | Viz.ai | Large vessel occlusion stroke detection on CT | De Novo | 2018 |
| Caption AI | Caption Health | AI-guided cardiac ultrasound for non-experts | De Novo | 2020 |
| Eko Analysis Software | Eko Health | Heart murmur detection from digital stethoscope | 510(k) | 2020 |
| Paige Prostate | Paige AI | Prostate cancer detection in biopsy slides | De Novo | 2021 |
| BriefCase Chest AI | Zebra Medical | Chest X-ray triage for critical findings | 510(k) | 2022 |
| Tempus ECG-AF | Tempus | Atrial fibrillation detection from 12-lead ECG | 510(k) | 2023 |
Key patterns: De Novo submissions were used for genuinely novel AI applications (first autonomous diagnostic AI, first AI-guided ultrasound). Once a De Novo is granted, subsequent similar devices use the 510(k) pathway with the De Novo as predicate. The trend is toward faster clearance times as the FDA builds institutional expertise with AI/ML devices.
Compliance Documentation Checklist

Use this checklist during development to ensure you are producing the required artifacts incrementally, not retroactively.
| Phase | Document | Contents | When to Produce |
|---|---|---|---|
| Planning | Intended Use Statement | Clinical purpose, target population, clinical setting, user profile | Before development begins |
| Planning | Risk Analysis (ISO 14971) | Hazard identification, severity/probability assessment, risk controls | Before development begins |
| Data | Data Management Plan | Sources, collection protocol, de-identification, labeling, quality criteria | Before data collection |
| Data | Dataset Description | Demographics, size, class distribution, feature statistics, split methodology | After data preparation |
| Development | Software Development Plan | Architecture, coding standards, version control, testing strategy | Start of development |
| Development | Algorithm Design Document | Model selection rationale, feature engineering, hyperparameter strategy | During model development |
| Validation | Validation Protocol | Test methodology, acceptance criteria, statistical analysis plan | Before validation testing |
| Validation | Validation Report | Results, subgroup analysis, calibration, comparison to predicate | After validation testing |
| Validation | Clinical Validation Report | Usability testing, human-AI team performance, clinical workflow analysis | After clinical testing |
| Submission | Model Card | Complete model documentation (see template above) | With submission |
| Submission | PCCP | Planned modifications, protocols, impact assessment | With submission (if applicable) |
| Post-Market | Monitoring Plan | Performance metrics, drift detection, complaint handling, reporting | Before deployment |
Frequently Asked Questions
Does my clinical decision support tool need FDA clearance?
It depends on the intended use and the degree of autonomy. Under the 21st Century Cures Act, CDS software that meets ALL four criteria is exempt from FDA oversight: (1) not intended to acquire, process, or analyze a medical image, signal, or pattern, (2) intended for the purpose of displaying, analyzing, or printing medical information, (3) intended for the purpose of supporting or providing recommendations to a healthcare professional, (4) intended to enable the healthcare professional to independently review the basis for the recommendation. If your CDS displays a risk score AND the underlying data/reasoning so the clinician can independently assess the recommendation, it may be exempt. If it provides an autonomous recommendation without transparency, it likely requires clearance.
How long does the 510(k) process take for AI/ML devices?
From submission to clearance, expect 90-150 days for a well-prepared 510(k) submission. The FDA has a 90-day review goal, but additional information requests (which occur in approximately 60% of AI/ML submissions) add time. The total calendar time from starting documentation to clearance is typically 6-12 months. De Novo submissions take longer: 150-300 days for review, with total timelines of 12-18 months.
Can we update our AI model without a new regulatory submission?
Yes, if you have an approved PCCP that covers the type of modification you are making. The PCCP must describe the modification type, the protocol for implementing it, and the validation criteria for accepting the update. Changes within the PCCP scope can proceed without a new submission. Changes outside the PCCP scope (new intended use, new patient population, architectural changes not anticipated in the PCCP) require a new submission.
What happens if our model degrades in production?
You have a legal obligation to monitor your device's performance and report certain events. Under the Medical Device Reporting (MDR) regulation, you must report to the FDA within 30 days if your device: (1) may have caused or contributed to a death or serious injury, or (2) has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur. Model degradation that leads to missed diagnoses or incorrect treatment recommendations may trigger MDR obligations. This is why drift monitoring is not just good engineering practice -- it is a regulatory requirement.
Do we need clinical trials for AI/ML devices?
Class II devices (510(k) and De Novo) typically do not require prospective clinical trials. Retrospective performance testing on labeled datasets is usually sufficient. However, the FDA may request a prospective study if: the device makes autonomous decisions (no clinician in the loop), the intended use involves high-risk clinical decisions, or the retrospective evidence is insufficient to demonstrate safety. Class III devices (PMA) always require prospective clinical trial data.
How does international regulation compare to FDA for AI/ML devices?
The EU's Medical Device Regulation (MDR 2017/745) classifies AI diagnostic software as Class IIa or IIb and requires CE marking through a Notified Body. The process is generally more burdensome than FDA 510(k) due to stricter clinical evidence requirements and the need for ongoing Notified Body oversight. The UK's MHRA is developing AI-specific guidance that is expected to align closely with the FDA's framework. Health Canada works jointly with the FDA on GMLP principles. For US-focused companies building healthcare software, start with FDA and use the documentation to accelerate international submissions.