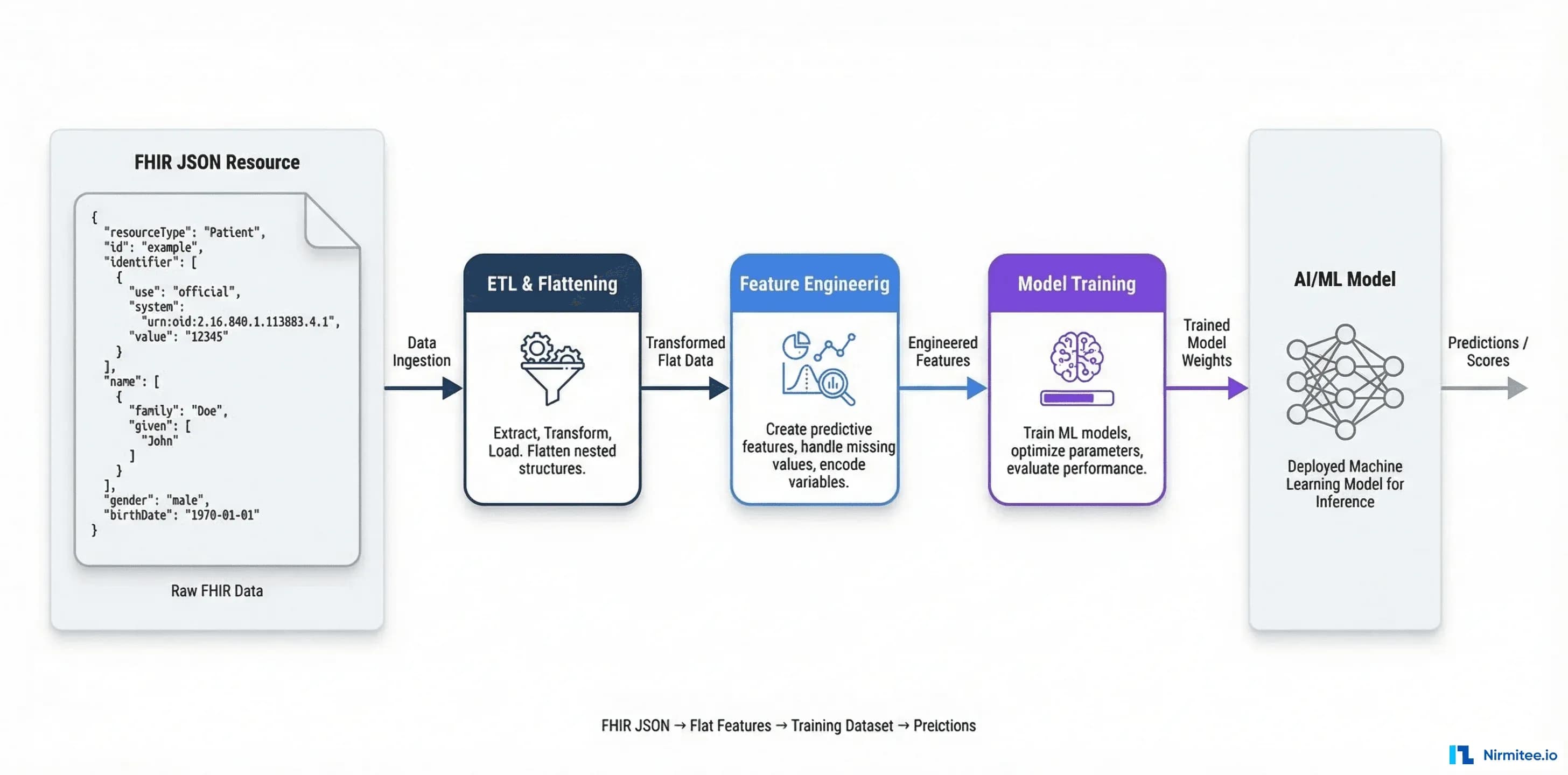
Why FHIR Data Is Hard for ML (And Why It's Still Your Best Option)
If you've ever tried to feed FHIR data into a machine learning pipeline, you've hit the wall. FHIR resources are deeply nested JSON documents with polymorphic fields, references to other resources via relative URLs, coded values from multiple vocabulary systems, and extension arrays that vary by implementation. A single Patient resource can contain nested arrays of identifiers, names (each with given/family sub-fields), addresses with period-of-use metadata, and contact points — none of which map cleanly to the flat feature vectors that ML models expect.
Here's what a typical FHIR Observation resource looks like — and why your pandas DataFrame doesn't know what to do with it:
{
"resourceType": "Observation",
"id": "blood-glucose-1",
"status": "final",
"category": [{
"coding": [{
"system": "http://terminology.hl7.org/CodeSystem/observation-category",
"code": "laboratory"
}]
}],
"code": {
"coding": [{
"system": "http://loinc.org",
"code": "2345-7",
"display": "Glucose [Mass/volume] in Serum or Plasma"
}]
},
"subject": {"reference": "Patient/patient-123"},
"effectiveDateTime": "2026-01-15T08:30:00Z",
"valueQuantity": {
"value": 126,
"unit": "mg/dL",
"system": "http://unitsofmeasure.org",
"code": "mg/dL"
}
}That's a single lab result. The value you actually care about for ML — 126 mg/dL — is buried four levels deep inside valueQuantity.value. The lab type is encoded as a LOINC code (2345-7) inside a nested coding array. The patient reference is a relative URL that requires a separate API call to resolve. And this is one of the simpler resources.
Despite this complexity, FHIR remains the best option for clinical ML pipelines for three reasons. First, standardization: unlike proprietary EHR exports (Epic's Caboodle, Cerner's HealtheIntent), FHIR resources follow a published specification. Code you write to parse FHIR Observations from one hospital works at another hospital. Second, regulatory momentum: the ONC Cures Act Final Rule mandates FHIR R4 APIs for certified EHR technology, meaning every major US health system now exposes FHIR endpoints. Third, bulk access: the FHIR Bulk Data Access specification ($export) provides a standard way to extract population-level data for analytics — exactly what ML training requires.
The engineering challenge isn't whether to use FHIR. It's how to transform FHIR's clinically-accurate-but-ML-hostile structure into training-ready datasets without losing the semantic richness that makes clinical data valuable. That's what this guide covers.
FHIR Resource to ML Feature: The Preprocessing Pipeline
The core challenge is flattening FHIR's hierarchical JSON into tabular features while preserving clinical meaning. Each resource type requires a different extraction strategy. Here's a production-tested approach for the four most common resource types in clinical ML.
Patient Demographics
Patient resources contain demographics that serve as foundational features — age, gender, geographic region. The complexity lies in FHIR's design: a patient can have multiple names (legal, maiden, nickname), multiple addresses (home, work, old), and identifiers from different systems.
import pandas as pd
from datetime import datetime, date
from dateutil.relativedelta import relativedelta
def extract_patient_features(patient_resource: dict) -> dict:
"""Extract ML features from a FHIR Patient resource."""
features = {}
# Age: calculate from birthDate
birth_date = patient_resource.get("birthDate")
if birth_date:
bd = datetime.strptime(birth_date, "%Y-%m-%d").date()
features["age"] = relativedelta(date.today(), bd).years
else:
features["age"] = None
# Gender: direct mapping
features["gender"] = patient_resource.get("gender") # male, female, other, unknown
# Geographic features: use the first 'home' address, fall back to any address
addresses = patient_resource.get("address", [])
home_addr = next((a for a in addresses if a.get("use") == "home"),
addresses[0] if addresses else {})
features["state"] = home_addr.get("state")
features["zip_3"] = (home_addr.get("postalCode") or "")[:3] # First 3 digits only (HIPAA safe)
# Marital status code
marital = patient_resource.get("maritalStatus", {})
marital_coding = marital.get("coding", [{}])[0]
features["marital_status"] = marital_coding.get("code")
# Active status
features["is_active"] = patient_resource.get("active", True)
# Identifier count (proxy for system engagement)
features["identifier_count"] = len(patient_resource.get("identifier", []))
return featuresKey design decisions: we extract only the first three digits of the zip code to maintain HIPAA Safe Harbor compliance. We use the home address preferentially. We calculate age dynamically rather than storing birth dates. These aren't just preprocessing steps — they're compliance requirements baked into the feature extraction layer.
Observations (Labs, Vitals)
Observations are where clinical ML gets interesting — and messy. A single patient might have thousands of Observations spanning lab results, vital signs, social history assessments, and survey responses. The challenge is pivoting time-series observations into patient-level features.
def extract_observation_features(observations: list, patient_id: str) -> dict:
"""Pivot FHIR Observations into patient-level features.
Handles: valueQuantity, valueCodeableConcept, valueString,
valueBoolean, and component (for blood pressure).
"""
features = {"patient_id": patient_id}
# Group observations by LOINC code
by_code = {}
for obs in observations:
if obs.get("status") in ("cancelled", "entered-in-error"):
continue
codings = obs.get("code", {}).get("coding", [])
loinc = next((c["code"] for c in codings
if c.get("system") == "http://loinc.org"), None)
if not loinc:
continue
effective = obs.get("effectiveDateTime", "")
value = None
# Handle polymorphic value[x] field
if "valueQuantity" in obs:
value = obs["valueQuantity"].get("value")
elif "valueCodeableConcept" in obs:
value = obs["valueCodeableConcept"].get("coding", [{}])[0].get("code")
elif "valueString" in obs:
value = obs["valueString"]
elif "valueBoolean" in obs:
value = 1 if obs["valueBoolean"] else 0
elif "component" in obs:
# Blood pressure: extract systolic and diastolic separately
for comp in obs["component"]:
comp_code = comp.get("code", {}).get("coding", [{}])[0].get("code")
comp_value = comp.get("valueQuantity", {}).get("value")
if comp_code and comp_value:
by_code.setdefault(comp_code, []).append((effective, comp_value))
continue
if value is not None and loinc:
by_code.setdefault(loinc, []).append((effective, value))
# Feature engineering: latest value, trend, count
LAB_MAP = {
"2345-7": "glucose", # Glucose
"2160-0": "creatinine", # Creatinine
"718-7": "hemoglobin", # Hemoglobin
"4548-4": "hba1c", # HbA1c
"2085-9": "hdl", # HDL Cholesterol
"2089-1": "ldl", # LDL Cholesterol
"8480-6": "bp_systolic", # Systolic BP
"8462-4": "bp_diastolic" # Diastolic BP
}
for loinc_code, feature_name in LAB_MAP.items():
values = by_code.get(loinc_code, [])
if values:
# Sort by date, take latest
sorted_vals = sorted(values, key=lambda x: x[0], reverse=True)
features[f"{feature_name}_latest"] = sorted_vals[0][1]
features[f"{feature_name}_count"] = len(sorted_vals)
# Trend: difference between latest and second-latest
if len(sorted_vals) >= 2:
latest = sorted_vals[0][1]
previous = sorted_vals[1][1]
if isinstance(latest, (int, float)) and isinstance(previous, (int, float)):
features[f"{feature_name}_trend"] = latest - previous
return featuresThis handles FHIR's polymorphic value[x] field — the same Observation resource might carry its value as a quantity, a coded concept, a string, or a boolean. Blood pressure is a special case: it uses the component array to store systolic and diastolic readings in a single resource. Missing these edge cases is how clinical ML models silently lose data.
Conditions (Diagnoses)
Conditions map to diagnosis features. The primary challenge is vocabulary: the same diagnosis might be coded in ICD-10-CM, SNOMED CT, or both. Your pipeline needs to handle all variants.
def extract_condition_features(conditions: list, patient_id: str) -> dict:
"""Extract diagnosis features from FHIR Condition resources."""
features = {"patient_id": patient_id}
# Track active conditions by ICD-10 chapter
icd10_chapters = {
"E": "endocrine", # E00-E89: Endocrine, nutritional, metabolic
"I": "circulatory", # I00-I99: Circulatory system
"J": "respiratory", # J00-J99: Respiratory system
"K": "digestive", # K00-K95: Digestive system
"M": "musculoskeletal", # M00-M99: Musculoskeletal
"N": "genitourinary", # N00-N99: Genitourinary
}
active_codes = set()
all_codes = set()
condition_count = 0
for cond in conditions:
# Skip refuted or entered-in-error
verification = cond.get("verificationStatus", {}).get("coding", [{}])[0].get("code")
if verification in ("refuted", "entered-in-error"):
continue
clinical_status = cond.get("clinicalStatus", {}).get("coding", [{}])[0].get("code")
is_active = clinical_status in ("active", "recurrence", "relapse")
codings = cond.get("code", {}).get("coding", [])
for coding in codings:
system = coding.get("system", "")
code = coding.get("code", "")
if "icd-10" in system.lower() or "icd10" in system.lower():
all_codes.add(code)
if is_active:
active_codes.add(code)
elif "snomed" in system.lower():
all_codes.add(f"snomed:{code}")
condition_count += 1
# Binary features for major disease categories
for prefix, category in icd10_chapters.items():
features[f"has_{category}"] = any(c.startswith(prefix) for c in active_codes)
# Common specific conditions
features["has_diabetes"] = any(c.startswith("E11") for c in active_codes)
features["has_hypertension"] = any(c.startswith("I10") for c in active_codes)
features["has_ckd"] = any(c.startswith("N18") for c in active_codes)
# Comorbidity burden
features["active_condition_count"] = len(active_codes)
features["total_condition_count"] = condition_count
return featuresMedicationRequests (Prescriptions)
Medication data reveals treatment patterns. The challenge: medications are coded using RxNorm, NDC, or proprietary formulary codes, and the dosage structure is deeply nested.
def extract_medication_features(med_requests: list, patient_id: str) -> dict:
"""Extract medication features from FHIR MedicationRequest resources."""
features = {"patient_id": patient_id}
active_meds = []
med_classes = set()
# RxNorm-based therapeutic class mapping (simplified)
DRUG_CLASSES = {
"metformin": "antidiabetic",
"insulin": "antidiabetic",
"lisinopril": "ace_inhibitor",
"amlodipine": "calcium_channel_blocker",
"atorvastatin": "statin",
"omeprazole": "ppi",
"metoprolol": "beta_blocker",
"levothyroxine": "thyroid",
}
for med in med_requests:
if med.get("status") not in ("active", "completed"):
continue
# Get medication display name
med_code = med.get("medicationCodeableConcept", {})
display = med_code.get("coding", [{}])[0].get("display", "").lower()
active_meds.append(display)
# Classify by therapeutic class
for drug_name, drug_class in DRUG_CLASSES.items():
if drug_name in display:
med_classes.add(drug_class)
features["active_med_count"] = len(active_meds)
features["on_antidiabetic"] = "antidiabetic" in med_classes
features["on_statin"] = "statin" in med_classes
features["on_ace_inhibitor"] = "ace_inhibitor" in med_classes
features["on_beta_blocker"] = "beta_blocker" in med_classes
features["polypharmacy"] = len(active_meds) >= 5 # Clinical threshold
return featuresCombining Into a Training DataFrame
With extraction functions for each resource type, building the complete dataset follows a straightforward pattern:
def build_patient_dataset(fhir_bundle: dict) -> pd.DataFrame:
"""Build a complete patient feature DataFrame from a FHIR Bundle."""
# Group resources by type and patient
resources_by_type = {}
for entry in fhir_bundle.get("entry", []):
resource = entry.get("resource", {})
rtype = resource.get("resourceType")
resources_by_type.setdefault(rtype, []).append(resource)
# Extract patient-level features
rows = []
for patient in resources_by_type.get("Patient", []):
pid = patient["id"]
# Demographics
features = extract_patient_features(patient)
features["patient_id"] = pid
# Observations for this patient
patient_obs = [o for o in resources_by_type.get("Observation", [])
if o.get("subject", {}).get("reference", "").endswith(pid)]
features.update(extract_observation_features(patient_obs, pid))
# Conditions
patient_conds = [c for c in resources_by_type.get("Condition", [])
if c.get("subject", {}).get("reference", "").endswith(pid)]
features.update(extract_condition_features(patient_conds, pid))
# Medications
patient_meds = [m for m in resources_by_type.get("MedicationRequest", [])
if m.get("subject", {}).get("reference", "").endswith(pid)]
features.update(extract_medication_features(patient_meds, pid))
rows.append(features)
df = pd.DataFrame(rows)
# Type conversions and encoding
bool_cols = [c for c in df.columns if c.startswith("has_") or c.startswith("on_") or c == "polypharmacy"]
for col in bool_cols:
df[col] = df[col].astype(int)
# One-hot encode gender
if "gender" in df.columns:
df = pd.get_dummies(df, columns=["gender"], prefix="gender")
return dfThis pattern — extract per resource type, merge on patient ID, encode for ML — is the fundamental building block of every FHIR-to-ML pipeline. Libraries like FHIRy and FHIR-PYrate automate parts of this, but for production pipelines, you'll want explicit control over feature engineering logic.
The Vocabulary Harmonization Problem
Your FHIR data arrives with codes from multiple vocabulary systems. The same clinical concept — "Type 2 Diabetes" — might appear as ICD-10-CM E11.9, SNOMED CT 44054006, or even a local code. If your ML model treats these as different features, it fragments the signal. Vocabulary harmonization is the process of mapping all codes to a unified representation.
ICD-10 vs SNOMED CT
ICD-10-CM is a classification system designed for billing and epidemiology. It groups diseases into categories. SNOMED CT is an ontology — it represents clinical meaning with hierarchical relationships. A SNOMED concept can map to multiple ICD-10 codes depending on clinical context. The NLM maintains an official SNOMED CT to ICD-10-CM map, but it's a many-to-many relationship that requires patient context (age, gender, comorbidities) to resolve correctly.
For ML purposes, the pragmatic approach is to standardize on one system per feature type. Use ICD-10 chapter-level groupings for comorbidity features (they're hierarchical by design), and SNOMED CT for clinical concept matching when you need semantic relationships.
LOINC for Labs
LOINC (Logical Observation Identifiers Names and Codes) is the standard for lab tests and clinical observations. It's well-structured for ML: each LOINC code maps to a specific test with defined units and specimen types. The challenge is that hospitals sometimes use local codes alongside LOINC, or use different LOINC codes for the same clinical test (e.g., glucose measured in serum vs plasma).
The LOINC-SNOMED CT extension (joint effort by Regenstrief Institute and SNOMED International) bridges lab terminology with clinical ontology, enabling queries like "find all observations related to diabetes monitoring" across vocabulary boundaries.
Building a Mapping Layer
A production vocabulary mapping layer needs to handle missing mappings gracefully, support multiple source systems, and be updateable as vocabularies evolve:
from typing import Optional, Dict, Set
import csv
from pathlib import Path
class VocabularyMapper:
"""Unified clinical vocabulary mapping for ML pipelines."""
def __init__(self, mapping_dir: str = "./vocab_maps"):
self.snomed_to_icd10: Dict[str, Set[str]] = {}
self.icd10_to_category: Dict[str, str] = {}
self.loinc_groups: Dict[str, str] = {} # LOINC code -> group name
self._load_maps(mapping_dir)
def _load_maps(self, mapping_dir: str):
"""Load NLM mapping files (download from UMLS)."""
map_path = Path(mapping_dir)
# SNOMED to ICD-10 (from NLM release)
snomed_file = map_path / "tls_Icd10cmHumanReadableMap.tsv"
if snomed_file.exists():
with open(snomed_file) as f:
reader = csv.DictReader(f, delimiter="\t")
for row in reader:
snomed_id = row.get("referencedComponentId", "")
icd10 = row.get("mapTarget", "")
if snomed_id and icd10:
self.snomed_to_icd10.setdefault(snomed_id, set()).add(icd10)
# LOINC groupings (from LOINC table release)
loinc_file = map_path / "loinc_groups.csv"
if loinc_file.exists():
with open(loinc_file) as f:
reader = csv.DictReader(f)
for row in reader:
self.loinc_groups[row["LOINC_NUM"]] = row["GROUP"]
def normalize_condition(self, codings: list) -> Optional[str]:
"""Normalize a condition's coding to ICD-10 chapter level."""
# Try ICD-10 first (preferred for ML categorization)
for coding in codings:
system = coding.get("system", "").lower()
code = coding.get("code", "")
if "icd-10" in system or "icd10" in system:
return code[:3] # Chapter-level grouping (e.g., E11 -> E11)
# Fall back: SNOMED -> ICD-10 via NLM map
for coding in codings:
if "snomed" in coding.get("system", "").lower():
snomed_code = coding["code"]
icd_codes = self.snomed_to_icd10.get(snomed_code, set())
if icd_codes:
return sorted(icd_codes)[0][:3] # Take first, chapter level
return None
def normalize_lab(self, codings: list) -> Optional[str]:
"""Normalize lab observation to LOINC group."""
for coding in codings:
if coding.get("system") == "http://loinc.org":
code = coding["code"]
return self.loinc_groups.get(code, code)
return NoneThis pattern lets you ingest FHIR data from any source — Epic, Cerner, open-source FHIR servers — and normalize to consistent features. The NLM mapping files are freely available through the UMLS licensing program.
Bulk FHIR Export ($export) for Training Data
Individual FHIR API calls (GET /Patient/123) are fine for clinical applications but terrible for ML. Training a model requires thousands to millions of records. The FHIR Bulk Data Access specification solves this with the $export operation — an asynchronous API that exports entire resource collections as NDJSON (newline-delimited JSON) files.
How $export Works
The workflow follows an async polling pattern:
# Step 1: Kick off the export
curl -X GET 'https://fhir-server.example.com/fhir/$export' \
-H 'Accept: application/fhir+json' \
-H 'Prefer: respond-async' \
-H 'Authorization: Bearer YOUR_TOKEN'
# Response: 202 Accepted
# Content-Location: https://fhir-server.example.com/fhir/bulkstatus/export-job-123
# Step 2: Poll for completion
curl -X GET 'https://fhir-server.example.com/fhir/bulkstatus/export-job-123' \
-H 'Authorization: Bearer YOUR_TOKEN'
# Response when complete: 200 OK with output file URLs
# {
# "transactionTime": "2026-03-14T10:00:00Z",
# "output": [
# {"type": "Patient", "url": "https://storage.example.com/export/Patient.ndjson"},
# {"type": "Observation", "url": "https://storage.example.com/export/Observation.ndjson"},
# {"type": "Condition", "url": "https://storage.example.com/export/Condition.ndjson"}
# ]
# }
# Step 3: Download NDJSON files
curl -o patients.ndjson 'https://storage.example.com/export/Patient.ndjson'Performance and Incremental Patterns
Full exports from large health systems can take hours and produce terabytes of data. Production pipelines use incremental exports with the _since parameter:
import requests
import ndjson
import time
from datetime import datetime
class BulkFHIRExporter:
"""Production bulk FHIR export client with incremental support."""
def __init__(self, base_url: str, token: str):
self.base_url = base_url.rstrip("/")
self.headers = {
"Authorization": f"Bearer {token}",
"Accept": "application/fhir+json",
"Prefer": "respond-async"
}
def start_export(self, resource_types: list = None,
since: str = None, group_id: str = None) -> str:
"""Initiate a bulk export. Returns the polling URL."""
if group_id:
url = f"{self.base_url}/Group/{group_id}/$export"
else:
url = f"{self.base_url}/$export"
params = {}
if resource_types:
params["_type"] = ",".join(resource_types)
if since:
params["_since"] = since # ISO 8601: "2026-03-01T00:00:00Z"
resp = requests.get(url, headers=self.headers, params=params)
resp.raise_for_status()
return resp.headers["Content-Location"]
def poll_until_complete(self, status_url: str,
poll_interval: int = 30,
max_wait: int = 3600) -> dict:
"""Poll export status until complete or timeout."""
elapsed = 0
while elapsed < max_wait:
resp = requests.get(status_url, headers=self.headers)
if resp.status_code == 200:
return resp.json() # Export complete
elif resp.status_code == 202:
# Still processing - check progress header
progress = resp.headers.get("X-Progress", "processing...")
print(f"Export in progress: {progress}")
time.sleep(poll_interval)
elapsed += poll_interval
else:
raise Exception(f"Export failed: {resp.status_code} {resp.text}")
raise TimeoutError(f"Export did not complete within {max_wait}s")
def download_ndjson(self, output_entry: dict) -> list:
"""Download and parse an NDJSON output file."""
resp = requests.get(output_entry["url"], headers=self.headers, stream=True)
resp.raise_for_status()
return ndjson.loads(resp.text)
# Usage: incremental daily export
exporter = BulkFHIRExporter("https://fhir.hospital.org/fhir", token="...")
status_url = exporter.start_export(
resource_types=["Patient", "Observation", "Condition", "MedicationRequest"],
since="2026-03-13T00:00:00Z" # Only data modified since yesterday
)
result = exporter.poll_until_complete(status_url)Security Considerations
Bulk exports contain PHI. Production deployments must encrypt data at rest and in transit, use short-lived download URLs (most implementations expire links within 1 hour), restrict exports to authorized backend services (not user-facing apps), and implement audit logging for every export operation. The Bulk Data Access IG v3.0.0 recommends SMART Backend Services authorization — machine-to-machine OAuth with signed JWTs — for export access.
Consent Management and HIPAA De-Identification
Using clinical data for ML isn't just a technical problem — it's a regulatory one. Every record in your training dataset represents a real patient, and you need both legal authority to use their data and technical safeguards to protect their identity.
Safe Harbor vs Expert Determination
HIPAA provides two methods for de-identifying protected health information (PHI):
Safe Harbor requires removing 18 specific identifiers: names, geographic data smaller than a state, dates (except year) for individuals over 89, phone numbers, fax numbers, email addresses, Social Security numbers, medical record numbers, health plan beneficiary numbers, account numbers, certificate/license numbers, vehicle identifiers, device identifiers, web URLs, IP addresses, biometric identifiers, full-face photographs, and any other unique identifying characteristic. It's deterministic and straightforward to implement — which makes it the standard choice for ML pipelines.
Expert Determination requires a qualified statistical expert to certify that the risk of re-identification is "very small." This preserves more data utility (you can keep dates, partial geographic data) but requires ongoing expert engagement and documentation. For ML pipelines where temporal features (admission dates, lab timing) are critical, Expert Determination may be worth the additional overhead.
FHIR Consent Resources
FHIR R4 includes a Consent resource that models patient consent decisions. For ML pipelines, this enables consent-aware data access — your ETL pipeline can check whether a patient has consented to research use before including their data in training sets:
def check_research_consent(consent_resources: list, patient_id: str) -> bool:
"""Check if a patient has active consent for research data use."""
for consent in consent_resources:
# Must reference this patient
patient_ref = consent.get("patient", {}).get("reference", "")
if not patient_ref.endswith(patient_id):
continue
# Must be active
if consent.get("status") != "active":
continue
# Check scope: research
scope_codings = consent.get("scope", {}).get("coding", [])
is_research = any(
c.get("code") == "research"
for c in scope_codings
)
# Check provision: permit or deny
provision = consent.get("provision", {})
provision_type = provision.get("type", "deny") # default deny
if is_research and provision_type == "permit":
return True
return FalseAudit Trails
Every access to clinical data for ML must be auditable. FHIR's AuditEvent resource provides a standard for logging data access. Production pipelines should create AuditEvent resources for every bulk export, every feature extraction run, and every model training job. When a patient revokes consent, your audit trail proves you can identify and purge their data from all downstream artifacts — a requirement under both HIPAA and GDPR.
Architecture: Production FHIR-to-ML Pipeline
Moving from notebook-level FHIR parsing to a production ML pipeline requires addressing data freshness, scalability, reproducibility, and compliance simultaneously. Here's the architecture that teams at Nirmitee have validated across healthcare AI implementations.
The Component Stack
Data Source Layer: The FHIR server (HAPI FHIR, Google Cloud Healthcare API, or Azure Health Data Services) is the source of truth. For organizations using openEHR as their clinical data repository, AQL (Archetype Query Language) provides an alternative query layer that's particularly powerful for analytics-oriented data extraction. AQL queries return tabular results directly — bypassing the FHIR-to-flat transformation step entirely.
-- openEHR AQL: Extract lab results as flat table directly
SELECT
e/ehr_id/value as patient_id,
o/data[at0001]/events[at0006]/data[at0003]/items[at0005]/value/magnitude as result_value,
o/data[at0001]/events[at0006]/data[at0003]/items[at0005]/value/units as result_units,
o/data[at0001]/events[at0006]/time/value as observation_time
FROM EHR e
CONTAINS COMPOSITION c
CONTAINS OBSERVATION o[openEHR-EHR-OBSERVATION.laboratory_test_result.v1]
WHERE o/data[at0001]/events[at0006]/data[at0003]/items[at0005]/name/value = 'HbA1c'
ORDER BY observation_time DESCETL Pipeline: Apache Spark or Python-based ETL (using frameworks like Prefect, Dagster, or Airflow) handles the heavy lifting: bulk export download, FHIR resource parsing, vocabulary normalization, feature extraction, and de-identification. For datasets under 1M patients, a well-optimized Python pipeline with multiprocessing handles the load. Beyond that, Spark's distributed processing becomes necessary.
Feature Store: A feature store (Feast, Tecton, or even a well-structured PostgreSQL schema) decouples feature computation from model training. Features computed once can be reused across models — the diabetes risk model and the readmission model both need the same HbA1c features. The store handles feature versioning, point-in-time correctness (avoiding data leakage from future observations), and serving features at inference time.
Model Training and Registry: MLflow or a similar registry tracks experiments, model versions, and the specific feature versions used for each training run. In healthcare, this lineage is critical: when a model is deployed clinically, you need to trace exactly which data, features, and code produced it.
Real-Time vs Batch
Most clinical ML use cases are batch: risk stratification scores computed nightly, cohort identification for clinical trials, population health analytics. These use bulk export + scheduled ETL. Real-time inference (e.g., clinical decision support during a patient encounter) requires a different pattern: FHIR Subscriptions or CDS Hooks to trigger feature computation on-demand, with pre-computed features from the store supplemented by real-time data from the current encounter.
The architecture should support both. Batch pipelines populate the feature store; real-time pipelines query it and augment with live data. This dual-path architecture is sometimes called the "lambda architecture" for clinical ML.
Tools and Frameworks
The ecosystem for FHIR-based ML has matured significantly. Here's what works in production today.
FHIR Servers
| Server | Bulk Export | Best For | Pricing |
|---|---|---|---|
| HAPI FHIR | Yes (v5.7+) | Self-hosted, full control | Open source (free) |
| Google Cloud Healthcare API | Yes | Managed, BigQuery integration | $0.03/10K FHIR ops |
| Azure Health Data Services | Yes | Microsoft ecosystem, DICOM + FHIR | $0.06/10K API calls |
| AWS HealthLake | Yes | AWS ecosystem, NLP built-in | $0.55/GB stored/month |
| Smile CDR | Yes | Enterprise, advanced access control | Commercial license |
Python Libraries
| Library | Purpose | Strengths |
|---|---|---|
| FHIRy | FHIR to DataFrame | Direct NDJSON/Bundle to pandas, simple API |
| FHIR-PYrate | FHIR query + extraction | Server querying, document filtering, DataFrame output |
| fhir.resources | FHIR data models | Pydantic models for validation and parsing |
| ML on FHIR | End-to-end ML | FHIR-native feature engineering, cohort definition |
| pandas + json_normalize | JSON flattening | Built-in, flexible, handles nested structures |
Data Processing
Apache Spark: The go-to for large-scale FHIR processing. The OHDSI Synthea ETL and Google FHIR Data Pipes projects provide Spark-based FHIR transformers. Spark handles the NDJSON files from bulk export natively and scales horizontally for multi-hospital datasets.
dbt (data build tool): For SQL-centric teams, dbt on top of a FHIR data warehouse (BigQuery, Snowflake) lets you define feature transformations as version-controlled SQL models. This is increasingly popular for analytics teams who aren't Python-first.
Great Expectations: Data quality validation for FHIR pipelines — catch missing codes, out-of-range lab values, and schema drift before they corrupt your training data.
Putting It All Together: A Practical Checklist
Building a clinical data pipeline that actually works — from FHIR resources to deployed ML model — requires getting the fundamentals right. Here's the decision framework:
- Data access: Use Bulk Data Export (
$export) for batch training. Use individual FHIR queries + EHR integration for real-time inference. If you have an openEHR CDR, AQL provides a direct path to flat analytics data. - Vocabulary normalization: Standardize on ICD-10 chapters for diagnoses, LOINC for labs, RxNorm for medications. Build the mapping layer once; reuse across all models.
- De-identification: Default to Safe Harbor for training data. Use Expert Determination only when temporal or geographic precision is critical to model performance.
- Feature engineering: Extract features per resource type, merge on patient ID. Handle FHIR's polymorphic value types explicitly. Validate feature distributions — clinical data has extreme outliers.
- Infrastructure: Feature store for reuse and point-in-time correctness. Model registry for lineage. Audit logging for every data access.
- Compliance integration: Build consent checking into the ETL pipeline, not as an afterthought. Check FHIR Consent resources before including patient data. Create AuditEvent resources for traceability.
The organizations building successful clinical AI — the ones getting past the 80% failure rate for healthcare AI projects — are the ones treating data engineering with the same rigor as model development. FHIR gives you a standardized starting point. The pipeline architecture described here gives you a proven path from raw clinical data to production ML. At Nirmitee, we build these FHIR and openEHR integration architectures for health systems that need their clinical data to power both care delivery and intelligent analytics — if you're tackling this problem, let's talk.
Struggling with healthcare data exchange? Our Healthcare Interoperability Solutions practice helps organizations connect clinical systems at scale. We also offer specialized Healthcare Software Product Development services. Talk to our team to get started.