"Does my app need FDA clearance?" That question is the $50,000 question every health AI startup asks. Getting it wrong in either direction is catastrophic. Over-classify, and you burn $500K-2M and 12-18 months on unnecessary submissions. Under-classify, and you face FDA warning letters, product recalls, and criminal liability.
On January 6, 2026, the FDA released revised CDS guidance that relaxed enforcement discretion for several AI-powered tool categories while drawing sharper lines around what remains regulated. This guide walks through every decision point — from first principles through risk classification and pathway selection. No ambiguity. No "consult your lawyer" hedging. Just the framework.
What Is Software as a Medical Device (SaMD)?
The IMDRF defines SaMD as software intended for medical purposes that performs those purposes without being part of a hardware medical device. Three distinctions matter:
- SaMD vs. SiMD: Software embedded in a scanner or pacemaker is SiMD — regulated as part of the hardware. SaMD runs independently on general-purpose hardware.
- SaMD vs. wellness: The software must claim to diagnose, treat, or prevent a disease — the statutory "medical device" definition. Fitness trackers and meditation apps are not SaMD.
- SaMD vs. administrative: EHR systems and billing tools handle medical data but have administrative intended use — not SaMD.
IEC 62304 and FDA's Regulatory Framework
If your software is classified as SaMD, compliance with IEC 62304 (Medical device software lifecycle processes) is mandatory for FDA submission, CE marking, and approval in virtually every major market. The standard requires documented requirements, architecture documentation, verification and validation at every lifecycle phase, risk management per ISO 14971, and maintenance planning. Its safety classification (Class A/B/C) maps to potential harm severity and dictates documentation rigor.
The FDA regulates SaMD under Section 201(h) of the FD&C Act. As of March 2026, the agency has authorized over 950 AI/ML-enabled medical devices, spanning radiology, cardiology, ophthalmology, oncology, and pathology. The pace has accelerated from 6 devices in 2015 to over 170 in 2023 alone.
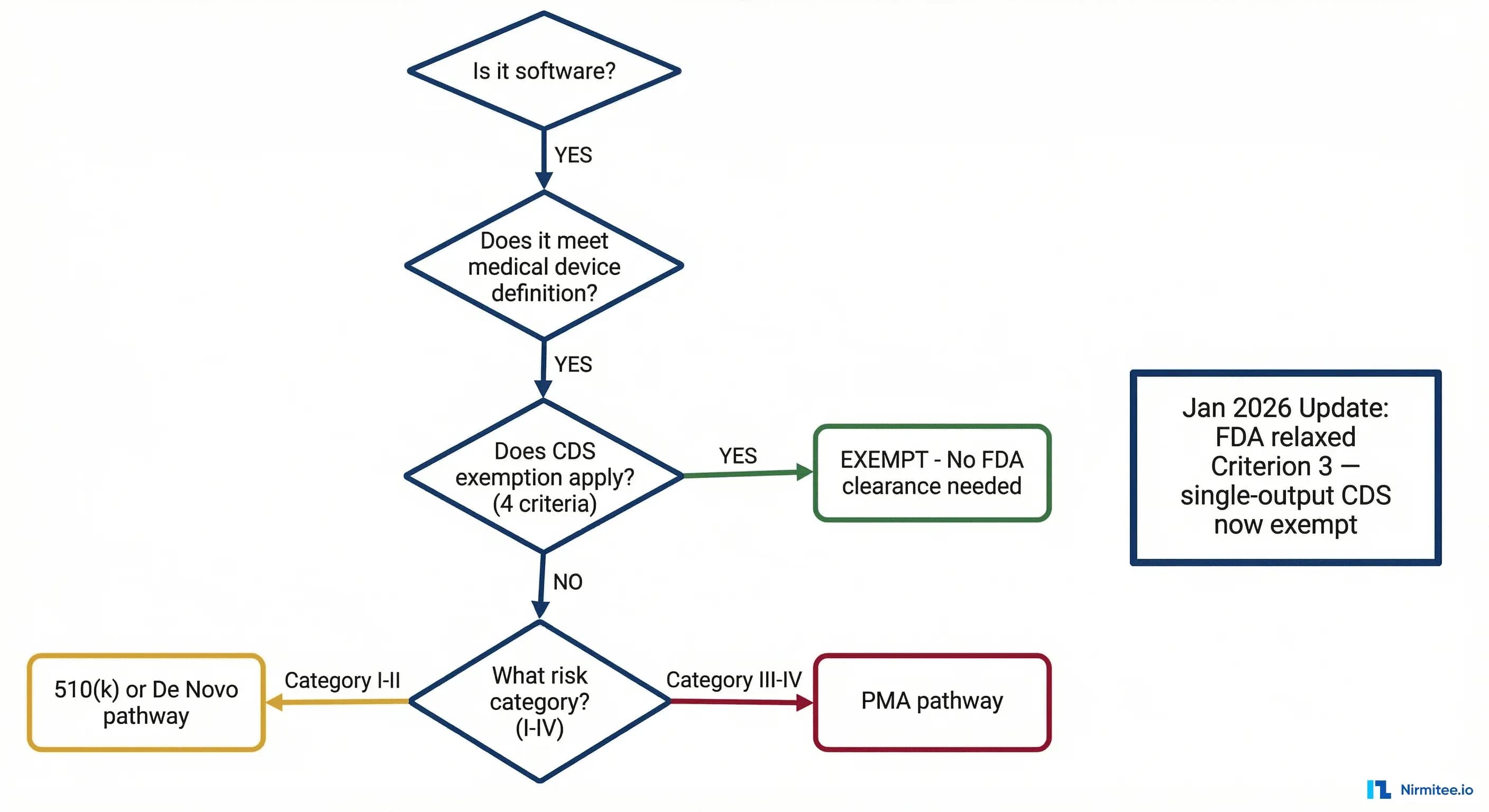
THE Decision Flowchart: Does Your Health AI Need FDA Clearance?
Five gates. Each is a binary decision. If your product exits at any gate, you have your answer.
Gate 1: Is It Software?
Question: Is your product software — defined as a set of instructions that processes input data to produce output?
- YES --> Proceed to Gate 2.
- NO --> Not SaMD. Not subject to SaMD regulation. (Example: a purely hardware sensor with no processing logic.)
If you are building a health AI application, your product is software. Move on.
Gate 2: Does It Meet the Medical Device Definition?
Question: Is the software intended to diagnose, treat, cure, mitigate, or prevent a disease or condition — OR is it intended to affect the structure or function of the body?
- YES --> Proceed to Gate 3.
- NO --> Not a medical device. Not regulated as SaMD.
Key nuance: Intended use is determined by your marketing claims, labeling, and promotional materials — not by what the software technically can do. A general-purpose LLM that could theoretically diagnose diseases is not a medical device if marketed as a general assistant. But the moment your website says "identifies early signs of diabetic retinopathy," you have established a medical intended use. General wellness apps, administrative tools, EHR systems, and education software typically fall outside the device definition.
Gate 3: Does the CDS Exemption Apply?
Question: Does your software meet ALL FOUR criteria for the Clinical Decision Support (CDS) exemption under Section 3060(a) of the 21st Century Cures Act?
This gate changed most significantly in January 2026. The four criteria are detailed in the next section.
- YES (all 4 criteria met) --> CDS-exempt. FDA exercises enforcement discretion. No premarket review required.
- NO (any criterion not met) --> Proceed to Gate 4.
Gate 4: What Is the SaMD Risk Category?
Question: Using the IMDRF risk classification framework (detailed in the Risk Matrix section below), what is the risk category of your SaMD?
The IMDRF framework produces four risk categories (I-IV) from two dimensions: healthcare situation severity and significance of the information provided.
- Category I (lowest risk) --> May not require premarket review (FDA enforcement discretion).
- Category II --> Likely requires 510(k) or De Novo.
- Category III --> Requires 510(k) or De Novo.
- Category IV (highest risk) --> Requires PMA (Premarket Approval).
Gate 5: Which Regulatory Pathway?
Question: Is there a substantially equivalent predicate device already cleared by the FDA?
- YES, predicate exists --> 510(k) pathway.
- NO predicate, but low-to-moderate risk --> De Novo classification request.
- High risk (Class III) --> PMA (Premarket Approval).
The remainder of this article unpacks each gate in detail.
The CDS Exemption: 4 Criteria (Updated January 2026)
Section 3060(a) of the 21st Century Cures Act carved out an exemption for Clinical Decision Support software that meets ALL FOUR of the following criteria. If your software satisfies every criterion, the FDA intends to exercise enforcement discretion — meaning it will not require premarket review.
Criterion 1: Not Intended to Acquire, Process, or Analyze a Medical Image, Signal, or Pattern
If your software takes in a chest X-ray, ECG waveform, pathology slide, dermoscopic image, or any other medical signal and processes it to produce a clinical output, Criterion 1 is not met. You are regulated.
Examples that FAIL Criterion 1:
- AI that reads chest X-rays for pneumothorax detection
- ECG analysis algorithms identifying atrial fibrillation
- Pathology image analysis for cancer grading
- Retinal image screening for diabetic retinopathy
Examples that PASS Criterion 1:
- Software that takes structured lab values (not raw signals) and computes a risk score
- CDS that flags drug-drug interactions based on medication lists
- Tools that analyze structured EHR data (vitals, diagnoses, demographics) for clinical alerts
Criterion 2: Intended for the Purpose of Displaying, Analyzing, or Printing Medical Information
The software must be intended to display, analyze, or print medical information about a patient or other medical information. This criterion is broadly inclusive — most CDS software meets it.
Criterion 3: Intended for the Purpose of Supporting or Providing Recommendations to a Healthcare Professional (HCP)
The output must be directed to an HCP — not directly to the patient for self-management of a serious condition. Software that provides diagnostic conclusions or treatment recommendations directly to patients without HCP intermediation does not meet this criterion.
January 2026 update: The revised guidance clarifies that single-output recommendations — risk scores, differential diagnosis lists, screening recommendations — qualify as "recommendations" to the HCP (not autonomous decisions), as long as the HCP can independently review the basis.
Criterion 4: Intended for the Purpose of Enabling the HCP to Independently Review the Basis for the Recommendation
This is the "transparency" criterion. The HCP must be able to understand WHY the software is making a recommendation — not just receive a black-box output. The software must provide the underlying data, the logic, or the reasoning chain that produced the recommendation.
This is where most AI/ML models face their hardest challenge. A deep learning model that outputs "87% probability of malignant melanoma" without any explainability layer fails Criterion 4. The clinician cannot independently review the basis — they can only accept or reject the output. For practical guidance on building explainability into clinical AI, see our deep dive on LLM explainability for healthcare clinicians and FDA compliance.
What satisfies Criterion 4:
- Displaying the input variables and their values alongside the recommendation
- Showing the clinical guidelines or evidence that informed the logic
- Providing feature importance scores, attention heatmaps, or SHAP values
- Referencing the specific published studies or clinical rules used
- Presenting the differential reasoning — why this recommendation over alternatives
January 2026 Guidance Changes: What Moved and Why It Matters
On January 6, 2026, the FDA issued revised final guidance titled "Clinical Decision Support Software." This was not a minor editorial update. It represented the most significant shift in CDS enforcement posture since the 21st Century Cures Act was signed in December 2016. Here is what changed.
Relaxation of Criterion 3: Single-Output Recommendations
The revised guidance explicitly extends enforcement discretion to CDS functions providing single-output recommendations: risk scores (sepsis, readmission, HEART, CHA2DS2-VASc), differential diagnoses, and screening recommendations. Previously, regulatory uncertainty about whether single outputs constituted "recommendations" or "autonomous decisions" led many startups to pursue 510(k) clearance defensively — burning $200K-500K and 6-12 months for products that now clearly fall within enforcement discretion.
Beyond Criterion 3, the guidance expanded enforcement discretion for laboratory analytics (structured lab trend analysis), medication management (dosing adjustments based on patient-specific factors), and care gap identification (overdue screening and vaccination alerts).
What Did NOT Relax
It is equally important to understand what the January 2026 update did not change:
- Image-based AI remains regulated (Criterion 1 unchanged).
- Patient-facing diagnostic tools remain regulated (Criterion 3 still requires HCP intermediation).
- Black-box AI remains regulated (Criterion 4 still requires explainability) — building guardrails should be a day-one priority.
- Generative AI clinical tools are under heightened scrutiny. The March 2026 breakthrough designation for a surgical AI chatbot confirms the FDA views these as regulated devices.
The Generative AI Signal
The FDA's March 2026 breakthrough device designation for a generative AI surgical chatbot signals two things: generative AI clinical tools are medical devices (not CDS-exempt), and the FDA wants to enable them through facilitative pathways with priority review and regulatory flexibility.
Many of the products navigating these classifications appear in our roundup of top healthcare apps leveraging GenAI.
Notably, FDA's own internal AI assistant "Elsa AI" switched from Claude to Gemini after hallucination issues — if the FDA itself struggles with LLM reliability, the bar for premarket submissions will be high. Understanding when rule engines outperform AI agents is not just an architecture decision — it is a regulatory strategy.
SaMD Risk Classification Matrix
Once you have determined that your software is SaMD and does not qualify for the CDS exemption, the next question is: how risky is it? The IMDRF developed a risk categorization framework that the FDA has adopted. It evaluates two dimensions:
- State of the healthcare situation: How serious is the patient's condition? (Non-serious, Serious, Critical)
- Significance of the information provided by the SaMD: What does the software do with clinical data? (Inform clinical management, Drive clinical management, Treat or diagnose)
The Classification Matrix
| Inform Clinical Management | Drive Clinical Management | Treat or Diagnose | |
|---|---|---|---|
| Non-serious condition | Category I | Category II | Category II |
| Serious condition | Category I | Category III | Category III |
| Critical condition | Category II | Category III | Category IV |
Understanding the Dimensions
Significance of information: "Inform" means the output supplements other information (e.g., population health dashboard). "Drive" means the output directly triggers a clinical decision (e.g., AI radiology report acted on directly). "Treat or Diagnose" means the software itself determines the diagnosis or treatment (e.g., closed-loop insulin dosing).
State of healthcare situation: "Non-serious" means no lasting harm from inaccuracy (e.g., acne assessment). "Serious" means potential irreversible harm (e.g., diabetes management). "Critical" means immediately life-threatening (e.g., stroke triage, sepsis detection).
Mapping Risk Categories to FDA Device Classes
The IMDRF categories do not map 1:1 to FDA device classes, but the general alignment is:
| IMDRF Category | Typical FDA Class | Regulatory Pathway | Examples |
|---|---|---|---|
| Category I | Class I (or enforcement discretion) | Exempt or 510(k) | Clinical data dashboards, non-critical trend analysis |
| Category II | Class II | 510(k) or De Novo | Radiology triage, risk stratification for non-critical conditions |
| Category III | Class II (higher risk) | 510(k) or De Novo | AI-assisted diagnosis for serious conditions, treatment recommendations |
| Category IV | Class III | PMA | Autonomous diagnosis of critical conditions, closed-loop therapeutic systems |
Regulatory Pathways Compared: 510(k) vs. De Novo vs. PMA
Once you know your risk category, you need to select the right regulatory pathway. The three primary options differ dramatically in cost, timeline, evidence requirements, and strategic implications.
The Comparison
| Dimension | 510(k) | De Novo | PMA |
|---|---|---|---|
| When to use | Predicate device exists | Novel, low-to-moderate risk | High-risk (Class III) |
| Total cost | $150K-500K | $300K-800K | $1M-5M+ |
| Timeline | 3-6 months | 6-12 months | 12-24 months |
| Evidence | Performance testing; clinical data sometimes | Performance + clinical validation; risk-benefit analysis | Prospective clinical trials |
| User fee (FY2026) | ~$22,000 | $0 | ~$440,000 |
| AI/ML share | ~85% | ~12% | <3% |
510(k): The Workhorse Pathway
The 510(k) accounts for ~85% of all AI-enabled device authorizations. The core requirement is demonstrating "substantial equivalence" to a predicate device with the same intended use and similar technological characteristics. Finding predicates has become easier as the installed base grows — radiology triage and ECG analysis now have dozens of predicates available. Clinical trials are not typically required; performance testing on curated datasets with demographic diversity and edge case coverage is usually sufficient.
De Novo: The Innovation Pathway
If no predicate exists and your device presents low-to-moderate risk, De Novo is the route. It has no user fee, creates a new regulatory classification (making your device the predicate for future competitors), and provides first-mover advantage. The tradeoff: 6-12 month timelines, a complete risk-benefit analysis, and the FDA may impose special controls binding on the entire device category.
PMA: The Clinical Trial Pathway
PMA is reserved for Class III (highest risk) devices — autonomous diagnostic or therapeutic systems for life-threatening conditions. Very few AI devices have gone through PMA. Expect prospective clinical trials, extensive safety monitoring, annual reporting, and $1M-5M+ in total costs over 12-24 months.
The Predetermined Change Control Plan (PCCP): Updating AI Models Without New Clearance
AI/ML models are not static. They are retrained, fine-tuned, and updated as new data becomes available. Under the traditional regulatory framework, every significant change to a cleared device requires a new premarket submission. For an AI model that might be retrained quarterly, this is untenable.
The FDA recognized this problem and introduced the Predetermined Change Control Plan (PCCP) framework — finalized in guidance in 2023 and now being implemented across AI/ML device submissions.
What Is a PCCP?
A PCCP is a pre-approved plan — submitted as part of your original 510(k), De Novo, or PMA application — that describes the specific types of changes you intend to make to your AI/ML device after clearance AND the methods you will use to manage the risks of those changes. If the FDA approves your PCCP, you can implement the described changes without filing a new premarket submission.
The Three Components of a PCCP
1. SaMD Pre-Specifications (SPS) — Describe the categories of changes you plan to make: retraining on new clinical sites, expanding patient populations, modifying architecture within defined parameters, updating training datasets, or adjusting sensitivity/specificity operating points.
2. Algorithm Change Protocol (ACP) — For each change category, define the testing protocol applied before deployment: performance benchmarks, test dataset requirements, statistical comparison methods, failure modes to monitor, and rollback criteria.
3. Impact Assessment — A risk analysis referencing your ISO 14971 file demonstrating that proposed changes maintain an acceptable risk-benefit profile.
Implementing a robust PCCP requires the kind of bounded autonomy architecture that constrains AI behavior within validated boundaries while enabling continuous improvement.
PCCP Strategic Considerations
- Scope broadly but credibly. Define change categories specific enough to be credible but broad enough to cover your product roadmap for 2-3 years. The FDA will push back on vague specifications.
- Invest in your ACP. Rigorous testing infrastructure, automated validation pipelines, and clear rollback procedures give the FDA confidence to approve a broad PCCP.
- Document everything. The FDA can audit your PCCP implementation at any time. Every model update needs a complete audit trail: ACP compliance, validation results, and risk impact assessment.
International Comparison: FDA vs. EU MDR vs. PMDA vs. Health Canada
Health AI is a global market. If you are building for US-only deployment, the FDA is your sole regulator. But most companies eventually expand internationally, and the regulatory requirements differ — sometimes significantly. In January 2026, the FDA and EMA aligned on 10 common principles for AI in medicine, signaling a move toward harmonization — but we are far from a single global standard.
| Dimension | FDA (US) | EU MDR / CE | PMDA (Japan) | Health Canada |
|---|---|---|---|---|
| Classification | Class I, II, III | Class I, IIa, IIb, III | Class I-IV | Class I-IV |
| AI/ML guidance | PCCP, AI/ML action plan, CDS guidance | MDCG 2019-11, AI Act (2025-2027) | MHLW guidance, IDATEN | ML pre-market guidance (2023) |
| Adaptive AI pathway | PCCP (pre-approved changes) | No equivalent; changes need new conformity assessment | IDATEN sandbox | Iterative updates with conditions |
| Clinical evidence | Performance testing; clinical trials for PMA | Clinical evaluation mandatory all classes | Varies by class | Proportionate to risk |
| Timeline | 3-24 months | 6-24 months | 6-18 months | 4-15 months |
Key Considerations
European companies face dual regulation: SaMD must comply with both EU MDR and the EU AI Act, which classifies most health AI as "high-risk" under Annex III with phased enforcement through 2027. The January 2026 FDA-EMA alignment on 10 guiding principles (data quality, transparency, human oversight, cybersecurity, equity) signals convergence but does not yet create mutual recognition — a device cleared by the FDA still requires independent CE marking.
Practical Compliance Checklist for Health AI Developers
Whether your product requires FDA clearance or qualifies for the CDS exemption, the following 15-item checklist represents the minimum viable compliance posture for a health AI company. Items 1-8 are essential even for CDS-exempt software. Items 9-15 are specific to products pursuing premarket review.
Foundation (All Health AI Products)
- Document your intended use statement. A single, unambiguous paragraph describing what your software does, for whom, and what clinical purpose it serves. This drives every downstream regulatory decision.
- Classify your product. Use the five-gate flowchart above. Document your rationale at each gate with specific evidence.
- Establish a Quality Management System (QMS). ISO 13485-aligned quality processes with document control, CAPA, complaint handling, and management review. Mandatory under 21 CFR 820 for regulated products.
- Implement risk management per ISO 14971. Hazard identification, risk estimation, control measures, and residual risk assessment — updated continuously.
- Build with IEC 62304 in mind. Structuring your SDLC around IEC 62304 from sprint one makes future regulatory pathways dramatically easier and cheaper.
- Address cybersecurity. The FDA's 2023 cybersecurity guidance requires SBOM, threat modeling, vulnerability disclosure, and patch management. For cloud SaMD, HIPAA-compliant architecture is the starting point.
- Ensure data quality and diversity. Document datasets: sources, demographics, size, labeling methodology, inter-annotator agreement, and known biases.
- Build explainability from day one. Black-box models are a liability. Invest in SHAP, LIME, attention visualization, and feature importance from the start.
Premarket Submission (Regulated SaMD Only)
- Conduct a predicate search. Use the FDA 510(k) database to identify substantially equivalent devices. No predicate? Evaluate De Novo.
- Prepare stand-alone validation testing. Test datasets must be independent of training data. Pre-specify success criteria for sensitivity, specificity, AUC, PPV, NPV with confidence intervals.
- Develop your PCCP early. Draft SPS + ACP in parallel with your initial submission. Align on scope via pre-submission meeting before filing.
- Maintain design controls per 21 CFR 820.30. Complete design history file: user needs, inputs, outputs, verification, validation, reviews, and transfer.
- Prepare labeling. Include intended use, performance data, training data characteristics, and known limitations. Be transparent about what the model cannot do.
- Plan post-market surveillance. Define complaint handling, adverse event reporting (MedWatch), performance monitoring, and real-world evidence collection before submission.
- Request a pre-submission (Q-Sub) meeting. Free, collaborative, and the single highest-ROI regulatory investment for first-time submitters.
Common Pitfalls That Delay or Derail Submissions
- Demographic imbalance in training data. The FDA has issued AI letters specifically citing inadequate demographic representation. Single-site or single-ethnicity datasets trigger pushback.
- Inadequate ground truth. Single-reader labeling without adjudication undermines validation methodology. Document annotator qualifications and inter-reader agreement.
- Overstating intended use. Marketing claims that exceed tested indications create an enforcement gap the FDA monitors actively.
- Ignoring edge cases. Document what happens with out-of-distribution inputs, degraded image quality, and unseen patient populations.
- No software lifecycle documentation. Retrofitting IEC 62304 compliance costs 3-5x more than building it in from sprint one.
Frequently Asked Questions
1. Is a chatbot that provides general health information considered SaMD?
It depends on intended use claims. General wellness information is not SaMD. Specific clinical recommendations ("you likely have condition X") meet the device definition. The March 2026 breakthrough designation for a generative AI surgical chatbot confirms clinically-specific chatbots are regulated. Operating within a structured AI maturity framework helps clarify boundaries.
2. Does the CDS exemption apply if the end user is a nurse or pharmacist?
Yes. The Cures Act refers to "healthcare professionals," not specifically physicians. Nurses, pharmacists, and PAs qualify for Criterion 3. The question is whether the output goes to a licensed professional who can independently evaluate it.
3. Does cloud deployment change the regulatory classification?
No. The FDA classifies SaMD based on intended use and risk, not deployment architecture. Cloud, on-premise, edge, or hybrid — the analysis is identical. Cloud does introduce additional cybersecurity and HIPAA considerations.
4. Could the FDA revoke the CDS exemption and make my product regulated?
The CDS guidance represents enforcement discretion, not a statutory exemption — so technically yes. Practically, the January 2026 revision expanded discretion (moving toward relaxation). Some companies pursue voluntary 510(k) clearance to eliminate this risk, particularly for enterprise health system sales where procurement requires FDA clearance.
5. How does the PCCP interact with annual reporting?
PCCP changes do not trigger new submissions but must be documented for FDA inspection and summarized in annual reports. The PCCP pre-authorizes change categories; it does not eliminate documentation requirements.
6. What is the fastest realistic path to FDA authorization?
Well-prepared 510(k) with clear predicate: 6-9 months (3 months prep + 3-6 months review). De Novo: 12-18 months. PMA: 24-36 months. First-time submitters should add 3-6 months for the learning curve.
Building for Regulatory Clarity
The regulatory landscape for health AI in 2026 is the most defined it has ever been. The January 2026 CDS guidance changes, the PCCP framework, the FDA-EMA alignment, and 950+ cleared AI/ML devices create a navigable environment for companies that invest in understanding it.
Three final recommendations:
- Classify early. Make the SaMD determination in your product definition phase — not after launch. Classification drives architecture (explainability), data strategy (diversity), and timeline expectations (3 months vs. 24 months).
- Build the quality system from day one. Retrofitting IEC 62304 is 3-5x more expensive than building it in. Every sprint should produce design history documentation, not just shipping code.
- Use the pre-submission meeting. The Q-Sub process is free, collaborative, and the single highest-ROI regulatory investment. No reason to skip it.
The health AI companies that will win the next decade are not the ones with the most sophisticated models. They are the ones that ship compliant products on predictable timelines. Regulatory fluency is not overhead — it is a competitive moat.
If you are building health AI products and need engineering teams that understand both the technology and the regulatory landscape, talk to Nirmitee. We build FHIR-native, compliance-ready healthcare software — from bounded autonomy architectures to real-time AI guardrail systems — for health AI companies that take regulatory seriously.